催化臭氧化吸附技術去除難降解污染物
李來勝,祝萬鵬,李中和
(清華大學環境科學與工程系,北京 100084)
摘 要:利用催化臭氧化吸附技術去除水溶液中溶解有機物的效率比單獨臭氧化或單獨吸附技術有明顯提高,它不但能將臭氧化難以降解的有機物氧化,而且還能減少后續氯化消毒工藝所形成的消毒副產物(三氯甲烷等),另外還可減少O3的用量且吸附劑一般不需再生。
關鍵詞:難降解污染物;消毒副產物;吸附;催化臭氧化
中圖分類號:TU991.2
文獻標識碼:B
文章編號:1000-4602(2002)05-0023-03
飲用水中的微量難降解污染物及消毒副產物需要用非生物降解處理技術去除,其中包括吸附和化學氧化法。
1 臭氧化與吸附技術
1.1 臭氧化
臭氧化反應機理為水中有機物可能直接與O3反應或與O3在水中分解產生的羥基自由基反應。雖然預臭氧化能去除一部分消毒副產物的前體物,但出水氯化后的致突活性與原水相比有較高的上升,另外O3僅是將對紫外光有較強吸收的大分子氧化成小分子,而不可能完全礦化為CO2和H2O,同時也產生一些副產物(如乙酸等),因此單獨使用O3并不是一種有效去除水中有機物的方法。
1.2 吸附技術
活性炭吸附是去除水中有機污染物最成熟有效的方法之一。活性炭可以去除水中致突物質,但它受吸附容量限制而不能有效去除氯化致突物的前體物;雖然它表面積大而且能用熱解析或在空氣中燃燒毒物的方法再生,但每次再生循環后都要損失一定量的活性炭。
某些無機材料由于具有吸附和催化的性質而引起廣泛關注,如MobilOil公司生產的M41S系 列新型無機中孔材料能被用作高容量吸附劑、仿生材料和氧化還原催化劑[1~4],其結構特征是表面積大、吸附力強(能吸附相當于其自身質量60%~70%的烴類),在去除水 中有機污染物方面具有很大潛力,表1中列出了幾種該系列產品的表面積[5、6]。Peter等發現鈦取代的MCM-41中孔分子篩對某些芳香烴類化合物及其衍生物的降解具有很 好的催化作用[6],可能具有非常廣闊的應用前景。
1.3 臭氧化與吸附技術結合
活性炭生物濾池的微生物活性常常有助于改善臭氧化對水中DOC的去除,這預示著臭氧化產生的化合物更易于生物降解。盡管生物過程使TOC去除效率增加,但活性炭對某些溶解有機物的吸附由于預臭氧化而惡化,因臭氧同腐殖質反應時產生更多活性炭不易吸附的極性化合物而導致活性炭吸附去除TOC性能下降。
鑒于在用鋁鹽混凝前水的預臭氧化能夠改善混凝時色度及有機物和三氯甲烷前體物的去除,因此推測比活性炭具有更多極性表面的吸附劑在預臭氧化后可能會比活性炭更有效地去除TOC。許多文獻報道活性氧化鋁吸附劑的吸附效率在預臭氧化后顯著改善,另外其他一些比活性炭具有較多極性表面的吸附劑(如鋁土礦、骨碳和活性氧化鋁)可能也有此效果。因此預臭氧化和氧化鋁或鋁土礦的吸附相結合可能是去除產生色度的有機物的優先選擇方法,但這還取決于吸附劑的再生能力及水溶液中有機物的特性。
2 催化臭氧化吸附技術
催化臭氧化技術是近年發展起來的一種新型的在常溫常壓下將那些難以用臭氧單獨氧化或降解的有機物氧化方法,同其他高級氧化技術(如O3/H2O2、UV/O3、UV/H2O2、UV/H2O2/O3、TiO2/UV和CWAO等)一樣,也是利用反應過程中產生的大量強氧化性自由基(羥基自由基)來氧化分解水中的有機物從而達到水質凈化。羥基自由基非常活潑,與大多數有機物反應時速率常數通常為106~109M-1·s-1,表2為某些有機物臭氧氧化與羥基自由基氧化速率比較。
從表2可見,羥基自由基與有機物反應的速率常數一般比臭氧的反應速率常數至少高出7個數量級。催化臭氧化吸附技術就是將催化劑的吸附技術與催化臭氧化技術相結合共同去除水中有機物。
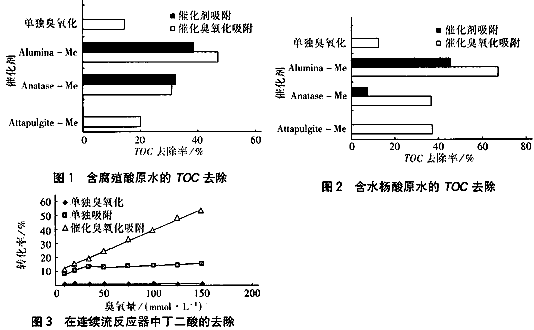
1999年法國的Legube B.和KarpelN.等研究了在銅系列催化劑作用下臭氧化對含有腐殖酸或水楊酸的飲用水的去除效果,發現其TOC去除率比同樣條件下單獨臭氧化有較大提高(模擬天然水:堿度=250mgCaCO3/L,pH=7.2,TOC=2.5~3.0mg/L,臭氧投加量為2.2~2.5mg/mgTOC),圖1、2分別為對含腐殖酸和水楊酸的原水進行催化臭氧化、催化劑吸附和單獨臭氧化去除TOC效率比較。
從圖1、2中可以看出,在沒有催化劑存在下對腐殖酸和水楊酸原水的TOC去除率分別為15%和12%;腐殖酸在三氧化二鋁(Alumina)基催化劑(300m2/g,10%Cu)、銳鈦礦(Anatase)基催化劑(61m2/g,5%Cu)和綠坡縷石(Attapulgite)基催化劑(59m2/g,5%Cu)的作用下,并未明顯改善TOC的去除率;但對水楊酸來說催化臭氧化比單獨臭氧化或單獨催化劑吸收去除TOC有明顯改善,三氧化二鋁基催化劑的催化臭氧化的TOC去除率(65%) 比單獨吸收(45%)和單獨臭氧化(12%)之和還要高,銳鈦礦和綠坡縷石基催化劑的催化臭氧化也是如此。同時他們還將金屬釕負載在二氧化鈰(200m2/g)上作催化劑氧化丁二酸(濃度=1mmol/L),丁二酸的降解程度隨臭氧量增加而直線增加(見圖3)。
Karpl N.等研究了過渡金屬催化臭氧化去除金屬成型設備沖洗循環水中的有機物、螯合劑和表面活性劑,圖4為含對甲苯磺酸溶液(TOC=8mg/L)在三種不同過渡金屬催化劑A、B和C作用下反應30min后TOC去除率的比較。
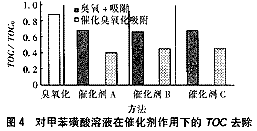
從圖4可以看出,在催化劑A作用下TOC去除率為60%,而同樣條件下先臭氧化后再用該催化劑吸附(去除溶液中的剩余臭氧后再吸附)和單獨臭氧化時TOC去除率分別為32%和12%。含特里通的原水在催化劑A作用下,催化臭氧化過程的TOC去除率(36%)較先臭氧化后再吸附(17%)和單獨臭氧化(10%)過程之和還要大得多。另外在催化劑A存在下,臭氧化對甲苯磺酸(反應30min)和3-硝基甲苯(反應10min)時消耗O3的量比先臭氧化后再吸附或單獨臭氧化要少。
西班牙的Gracia在研究EBRO河水處理過程中發現在催化劑TiO2/Al2O3(132.5m2/g)存在條件下,催化臭氧化后再氯化產生的三鹵甲烷(THMs)量較預臭氧化或預氯化后再氯化所產生的THMs少且所產生的THMs濃度隨著臭氧劑量的增加而減少,其數據見表3。?
6
24 0.4
0.3
0.1 1
5
24 6
14
39 6
12
16 0
0
1 13
31
80 單獨臭氧(1.6gO3/gTOC) 2
6
24 0.5
0.3
0.1 1
7
23 5
11
30 4
8
21 0
0
1 10
26
75 催化臭氧化(0.8gO3/gTOC) 2
6
24 0.4
0.3
0.1 1
2
25 5
8
30 4
7
21 0
0
1 10
17
77 催化臭氧化(1.6gO3/gTOC) 2
6
24 0.5
0.3
0.1 1
2
25 3
8
26 2
6
11 0
0
0 6
16
62 預氯化預氯化加后氯化 24
2
6
24 0
0.7
0.5
0.2 3
15
34
55 4
16
27
30 2
13
17
23 0
0
0
1 9
44
78
108 注:TOC=4.46 mg/L,投氯量為2.5 mg/L。
Volk C.等發現在后續氯化消毒處理過程中,催化臭氧化預處理較同樣條件下單獨臭氧化或臭氧過氧化氫氧化所需的氯量減少,從而使消毒副產物大為減少。
3 結語
催化臭氧化對水中有機物去除率較單獨吸附和單獨臭氧化之和還要高,而且消耗的臭氧量也大為減少;氯化消毒處理時,催化臭氧化比同樣條件下單獨臭氧化或臭氧過氧化氫氧化所需的氯量減少;此外即使在氯化消毒工藝加同樣的氯量,催化臭氧化作為氯化預處理工藝所產生的三鹵甲烷量較預臭氧化和預氯化工藝所形成的三鹵甲烷量少。因此如果能夠找到一種吸附表面積較大的催化劑(如MCM-41),那么就能利用催化劑的巨大表面積和對化學物質的選擇吸附性,將有機物和氧化劑同時吸附到催化劑表面,在吸附劑表面形成高濃度有機物和氧化劑,使液相催化氧化反應轉化為固相表面上的催化氧化反應,這樣不但可以加快氧化反應速度而且可有選擇地氧化,在常溫常壓下將水中難降解的有機物更大程度氧化或降解,同時可減少氧化劑用量以及三鹵甲烷的產生量。另外,該類吸附劑基本上不需要再生。
參考文獻
[1] Huo Q,Margolese D I,Ciesla V,?et al.Generalised synthesis of periodic surfac tant/inorganic composite materials[J].Nature,1994,368:317-319.
[2]Inagaki S,Fukushima Y,Kuroda K.Synthesis of highly ordered mesoporous mat erials from a layered polysilicate[J].J Chem Soc Chem Comm,1993,3:680-682.
[3]Monnier A,Schuth F,Huo Q,?et al?.Cooperative formation of inorganic-organic in terf aces in the synthesis of silicate mesostructures[J].Science,1993,261:1299-1301.
[4]Rathousky J,Zukal A.Adsorption on MCM-41 mesoporous molecular sieves:Part 2.Cycl opentane isotherms and their temperture dependence[J].Chem Soc Faraday Trans,1991,91(5):937-940.
[5]Coolin Cooper,Robbie Burch.Mesoporous materials for water treatment processes[J].Water Research,1999,33(18):3689-3694.
[6]Peter T Tanev,Malama Chibwa,Thomas J Pinnvaia,?et al.Tianium-containing mesop orous molecular sieves for catalytic oxidation of aromatic compounds[J].Nature ,1994,368:321-323.
電 話:(010)62784527?
E-mail:questers@99mails,tsinghua.edu.com
收稿日期:2001-11-27
論文搜索
月熱點論文
論文投稿
很多時候您的文章總是無緣變成鉛字。研究做到關鍵時,試驗有了起色時,是不是想和同行探討一下,工作中有了心得,您是不是很想與人分享,那么不要只是默默工作了,寫下來吧!投稿時,請以附件形式發至 paper@h2o-china.com ,請注明論文投稿。一旦采用,我們會為您增加100枚金幣。



